1. Introduction to the Investment Thesis
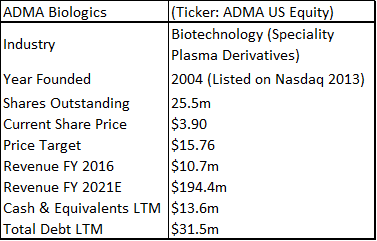
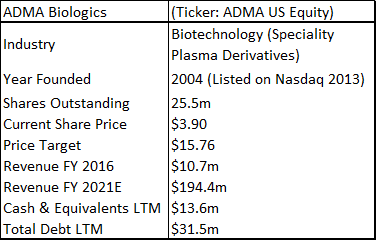
ADMA Biologics is a sub-$100m capitalization healthcare company that is based in Boca Raton, Florida. The company is aiming to commercialize a novel high-titer hyper-immune globulin product for the treatment of RSV and related respiratory viruses in immune compromised patients. ADMA listed as a public company in February 2013 (on the Nasdaq OTC market through a reverse merger).
The two main contentions of this positive investment thesis are; 1), the burden of lifting the regulatory hold at the Boca Raton facility, while being a significant challenge for management, should prove to be within reach; 2), The long-term potential of entering the plasma market as a small supplier of niche high-value hyperimmune products is a very attractive business proposition for ADMA. While it is unusual for large and strategically important fractionation facilities to experience regulatory issues, the overall regulatory landscape pertaining to plasma processing, fractionation and logistics is clearly defined by the relevant regulatory authorities.
ADMA has had three capital injections since it took control of the Boca facility. It obtained additional financing as part of the BTBU transaction, it refinanced its debt with Marathon Asset Management, and it raised $39.1m in additional equity. ADMA management is now confident that it will have sufficient cash resources to progress the company into the first half of 2018, in advance of key corporate milestones. ADMA’s current cash burn rate is c.$3m to c.$3.5m per month, and the company ended Q3 2017 with c.$13m in cash. Revenues for the nine months of 2017 were $10.8m. Through the transaction agreement, ADMA secured a ten-year plasma supply agreement with Biotest AG (and/or its successors), along with a three-year post-closing agreement, for the purchase of RSV plasma from the two collection centers, at a price equal to cost plus 5% (excl-inflation). Biotest AG is currently engaged in a potential buy-out process with Chinese private equity firm Creat Group (an affiliate of Shanghai RAAS, the China based pharmaceutical conglomerate). ADMA appears to be stuck between a rock and a hard place. The company is burning its cash resources and appears to be stuck in regulatory limbo. However, upon detailed analysis it is clear that these concerns are overblown.
If ADMA can pass an FDA cGMP re-inspection at the first time of asking, it will be in a much stronger strategic position relative to when it first became a public company. Currently the market isn’t willing to ascribe any ‘benefit of the doubt‘ valuation on the shares, and the company is being valued as an entity with considerable capital but with few valuable assets. From the perspective of the long-term healthcare investor, ADMA shares now represent a very favorable investment opportunity. The company is ‘off the radar‘ for the average investor, but still possesses a relatively liquid share float and has a listing on the Nasdaq. Perhaps the most interesting aspect surrounding ADMA’s current valuation, is the lack of value that ADMA’s current share price places on RI-002 (especially in light of the current overvaluation in the biotechnology industry). ADMA has generated strong phase III data (in fact the drug produced some of the strongest clinical results ever for an IVIG). The drug has a modest hurdle for approval and there is a significant unmet medical need among immune compromised (IC) patients, who are vulnerable to developing RSV and other common respiratory viruses. ADMA’s approach of isolating high-titer antibodies to specific infections is a novel one, with the larger plasma company’s focusing on recombinant technologies, hemophilia and breakthrough vaccines for rare diseases.
2. 2018 is the make-or-break year for ADMA
The upcoming FDA re-inspection at the Boca facility is the key business milestone for ADMA. Management has guided that the facility will be ready for re-inspection in late 2017.
Figure 2.1: Anticipated milestones in Q4 2017 and beyond
Source: Analyst Estimates
3. The BTBU transaction has transformed ADMA strategic positioning
The fractionation facility that ADMA has acquired is 126,000 sq.m in size, and has a peak capacity of 400,000L per annum. The site also includes a state-of-the-art monoclonal antibody production facility, along with 15 acres of land adjoining the buildings. The facility employed c.900 employees and generated total revenues of $76.5m in FY2016.
Figure 3.1: The BTBU generated $76.5m in revenues for FY 2016

Source: ADMA Biologics Prem. Form 14A
Biotest originally purchased the Florida facility from Nabi Pharmaceuticals in 2007, (Nabi had built the facility in 2001) for a transaction value of $185m (which incl. 9 plasma collection centers). Biotest invested $50m in upgrading and expanding the facility and invested $70m in constructing an adjoining state-of-the-art monoclonal antibody production unit. The transaction should be highly accretive for new shareholders. With vertical integration, management have guided that a blended gross profit margin ranging c.60% to 70% is achievable (prior guidance was for top-line margins of c.40% to 50%).
4. Primary Immune Deficiency and the adaptive immune system
Primary immune deficiency disorder is the collective term for a range of medical disorders that occur when the adaptive immune system is malfunctioning. Patients with one or more forms of PIDD, often have defects in their t-cells or b-cells (important for the immune system’s ability to adapt to the presence of foreign pathogens) as opposed to defects in the innate immune system (phagocyte and complement disorders). There are relatively concrete population prevalence estimates, that can be gathered from a range of studies conducted in hospitals across the United States. These place the total US patient population, at approximately 1 in 1,200, or 250,000 people.
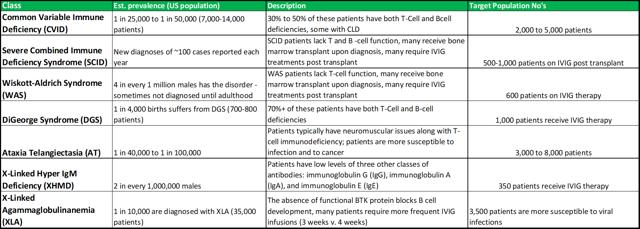
PIDD disorders are commonly viewed by physicians as falling into the rare diseases category. The exact prevalence is difficult to estimate, as there are no standard screening procedures for immune system defects in the United States (or elsewhere in the world). The main disorders that fall under the PIDD category are listed in figure 4.1 (below).
Figure 4.1: ADMA intends to focus on the high-risk segments within the PIDD patient population
Source: ADMA Biologics
IVIG has been the global standard of care for PIDD patient’s in recent decades, and IVIG treatments are still viewed by physicians as the most appropriate treatment option for PIDD. IVIG’s are a powerful cocktail of immune system antibodies, which are isolated from pools of plasma collected from thousands of plasma donors. The concept of immune globulin use has a long history across the medical landscape and has been widely indicated for use in passive immunization. In these instances there is a high risk of infection and the body has insufficient time to develop its own immune response, or to reduce the symptoms of ongoing or immunosuppressive diseases. While the functioning of the immune system is well characterized, it has proven very difficult to replicate the near natural effects of immune system supplementation that can be achieved through IVIG treatment (to date chemical and monoclonal antibody formulations have not achieved similar effects). The increasing use of self-administered solutions, which allow patients to treat their conditions in an out-patient setting, has contributed to the predominant position that IVIG now occupies. IVIG use is particularly beneficial among young children and babies which have under-developed immune systems. Infants have particular safety and efficacy needs which often can’t be accommodated through treatment with standard vaccines.
On the other hand, IVIG’s have unique treatment profiles. As the formulations are largely comprised of the natural human antibodies, their safety profile is limited to complications that might arise during the manufacturing process, or with complications that might be observed among plasma donors, or with complications that might be observed during the administration of the final product. IVIG’s can be combined with other therapies, including new therapies that are introduced onto the market, without having to demonstrate efficacy or drug-drug interactions, etc. IgG can be modified for the use of MaB’s and used as a carrier to enhance therapeutic efficacy.
When side effects do result from IVIG use, they have resulted from the tendency of IgG to form aggregates or clots when the plasma is purified during the manufacturing stage. This is often caused by filter clogging. If a patient is administered a solution that contains coagulants it can cause potentially severe reactions. Complications can also arise from excessive donation and this has been a focal point for the anti-donation lobby (more discussion in Section 17 below). The physical effects experienced during a donation session are not dissimilar to those experienced while donating blood, e.g., mild injection site reactions, feelings of nausea, etc., although reaction rates have been reported to be higher for blood donations. The frequency of acute reactions among donors undergoing apheresis procedures was found to be 2.18% in a multi-institutional study. In comparison, in a review of re actions among whole blood donors, an overall reaction rate to whole blood donation of 11 to 21% was reported. A 2005 study found hypocalcemia to be the most common aphaeresis-specific reaction (caused by citrate anticoagulation). Further study is needed to determine the effect this might have on the long-term plasma donor. Hypocalcemia is caused by the excess presence of sodium citrate in the blood stream. This bonds with calcium in the blood, which in turn leads to reduced circulating calcium levels, and in extreme cases can result in hypocalcemia. In non-extreme cases donors can experience fainting, tingling, numbness, muscle contractions or seizures. Bone demineralization can also result from long-term decalcification. In the PIDD subset of IC patients, c.50% of the US population is actively using an IVIG formulation.
RSV has an infection rate of c.5%-15% in the IC population (off-label use is widespread). ADMA contends that physicians will be receptive to using a proven medical solution to treat the full range of IC patients, who are vulnerable to contracting respiratory viruses. It is widely understood that patients are often misdiagnosed and/or left undiagnosed for a number of years after the initial presentation of symptoms. The American Academy of Pediatrics (in its bronchiolitis management guidelines), does not specifically recommend RSV testing. This increases the risk for recurrent upper and lower respiratory tract infections, as well as for bacterial sepsis, which can be immediately fatal.
Reverse transcription polymerase chain reaction is an expensive assay testing technique that is not commonly used outside of the emergency department setting, owing to the expense and lack of available results while the child is in the ED. A study of US children under the age of 5 years who were tested for RSV independent of their discharge diagnosis, showed that only 45% were diagnosed with an RSV-associated illness, while in the outpatient setting RSV positive children had a 3% RSV diagnosis. RSV is commonly misdiagnosed as otitis media, dyspnea, wheezing, and croup.
Patients who continually suffer recurrent respiratory infections are placed at an increased risk of suffering chronic lung disease and bronchiectasis. The benefit of therapeutic immunoglobulin use in this population is seen in its ability to reduce the incidence rate of infections. This has been attributed to the diverse antibody specificities against common viral pathogens present in plasma formulations. While the FDA requires IVIG products to pass potency tests for minimal levels of antibodies against measles, diphtheria, and polio, these pathogens do not commonly result in infections among PIDD patients. Currently available IVIG products are not standardized to contain specific antibody content to defend against the most common infectious diseases that PIDD patients experience. Studies of the available products have reported a wide variation in antibody titres.
When ADMA is able to market its full stable of products, it can focus its sales effort for Bivigam towards the general PIDD population, while RI-002 can be focused towards higher-risk patients. The total addressable market for RI-002 could be c.8,500 to c.15,000 patients. The high-risk/rare disease nature of these patients, should allow RI-002 to command a premium price, compensating for the relatively small total addressable market. ADMA intends to put RI-002 forward for initial approval, with a label indicating treatment for a range of respiratory viruses that are commonly observed among PIDD patients. In light of RI-002’s positive phase III results and its strong effect on RSV antibody elevation, it has strong potential to be viewed by physicians as a core treatment option for RSV infection during the winter months (complementary potency against other common respiratory viruses should be viewed very favorably). RI-002 won’t be contraindicated with other treatments commonly used by these patients (distinguishing it from vaccines).
Following approval, ADMA can pursue additional indications to treat patients who are vulnerable to contracting RSV (and other common respiratory viruses) due to their IC status. A particularly large group that could benefit from RI-002 treatment, are patients undergoing chemotherapy treatment. These patients are placed at an elevated risk of mortality after contracting viral infections. An important benefit of antibody treatment is that it can be synergistic or additive when combined with conventional antimicrobial chemotherapy against bacterial fungal and viral infections. RI-002 was probably renamed (originally known as RI-001 before management communicated introduction of minor changes in production process) to maintain clinical profile comparability and smooth the process of label expansion post-approval. For plasma derivatives comparability guides the development process during the preclinical phases. This ensures that clinical data is linked to the product intended for commercialization. A robust comparability program during all stages of the product life-cycle shortens the time to licensure and facilitates the implementation of post approval changes. The CBER’s plasma products paradigm is that the manufacturing process is the product that is ultimately approved and licensed.
Figure 4.2: IVIG is widely used both on-label and off-label

Source: FDA & CBER.
5. RI-002’s phase III results “batted the ball from the park”
RI-002 demonstrated an incidence rate of zero SBI’s in 55.88 patient years of treatment, which resulted in an SBI rate below the FDA’s criteria of <1.0 SBI per subject per year. Other key secondary endpoints were well within a reasonable range expected to observed in IVIG's (see figure 6.1 below). A difference in specific antibody levels was observed among patients who received doses ?500 mg/kg in comparison to patients who received doses ?500 mg/kg. Figure 5.1: RI-002 demonstrated zero SBI’s and a very low rate of hospitalizations due to infection

Source: Wasserman et al.
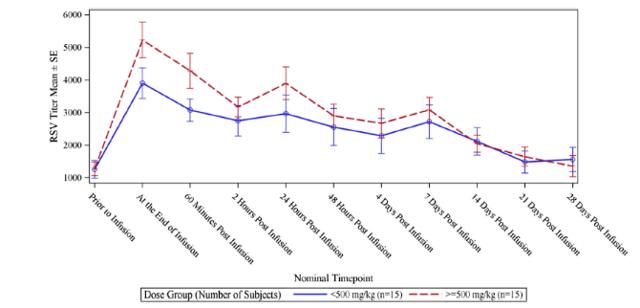
RI-002 administration produced increases in all of the studied antibodies, with the greatest increase observed in antibodies to RSV, with a 5.47-fold increase in titres after infusion of the IVIG and a 6.79-fold increase in those subjects who received greater than 500 mg/kg (figure 6.2 below). The applied baseline measurement was the value of anti-RSV measured immediately prior to trial entry. Figure 6.3 (below) shows the PK of anti-RSV antibodies, where the baseline was the value of anti-RSV measured in subjects enrolled in this portion of the study, immediately prior to infusion of the seventh or ninth dose of IVIG.
Figure 5.2: Mean IgG plasma concentration levels and mean Cmax values were similar across treatment groups despite the slightly longer treatment period
Source: Wasserman et al.
Figure 5.3: The dosage schedule produced significantly elevated levels of RSV antibodies (from baseline) in both treatment groups

Source: Wasserman et al.
RI-002 was safe and efficacious and the portion of study infusions that experienced a TAAE was 0.142, with an upper one sided 95% confidence limit of 0.164, which was well within the target safety endpoint of (<0.40) (figure 6.4 below documents the adverse events schedules). Figure 5.4: Summary of temporally associated and treatment emergent adverse events


Source: Wasserman et al.
During the study, 43 (72.9 %) of subjects experienced at least one TAAE, and TAAE’s occurred during 113 (14.2 %) of study infusions. The mean (SE) planned weight adjusted administered dose of RI-002 was 505.2 (4.84) mg/kg. Study infusions were well tolerated with 95.8% of infusions administered at the maximum allowed infusion rate of 8 mg/kg/min. The use of pre-medication was limited to seven study infusions (from a total of 793 infusions) in seven subjects (0.009%). Of the 618 TEAE’s that were experienced by 58 subjects, the most common were headache, sinusitis diarrhea, viral gastroenteritis, nasopharyngitis, and upper respiratory tract infection (common illnesses in this population). There were no reported serious adverse events related to RI-002, and reported the two events, were experienced as a single event in a single subject.
Figure 5.5: Total IgG PK parameter estimates show well-expressed concentration profiles between subjects in both treatment cycles
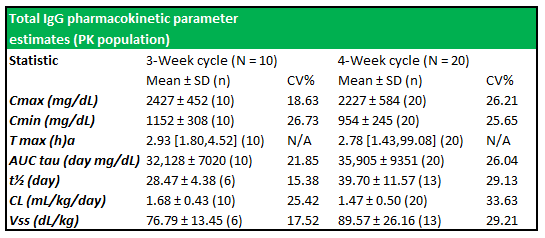
Source: Wasserman et al.
6. ADMA’s trip to regulatory purgatory should be short lived
Two important points to consider when assessing the regulatory burden that ADMA faces at the Boca facility; are 1), it is very unusual for fractionation facilities to experience regulatory issues (several industry representatives expressed this view in telephone conversations); 2), the regulatory burden for plasma fractionators is very well defined in the relevant regulatory statutes; 3) industry practices and standards are well-established and there is a considerable industry expert body-of-knowledge in relation to these specific issues. This has built continually built upon over decades of practice in the United States. Speculating as to the reasons why the facility has experienced difficulties over a prolonged period, is not important in the assessment of ADMA’s ability to remediate the issues at the facility. ADMA has communicated with investors that the FDA regional inspectors took issue with BTBU responses to questions such as, “what would you do if something went amiss with such-and-such a process“. Management outlined that BTBU’s provision of clear operating procedures and staff training protocols was below regulatory standards. ADMA feels that if management are more critical of out-of-specification operational and manufacturing processes, and develop procedures that are less cumbersome and easier to follow, they will be able to operate the facility within the regulatory framework. A central aspect of preparing the facility for a re-inspection will be the guidance that it receives from Validation Consulting. Engaging external subject matter experts when changing a manufacturing process is common practice in the fractionation industry. BTBU didn’t appear to use this approach when it attempted to change aspects of the processes used to manufacture Bivigam (more details on this aspect below). It is also possible that managing the adjoining MaB bioreactor facility, was an undue burden on BTBU management. Three MaB’s were progressed through a clinical development program but were subsequently discontinued.









Leave A Comment